
One way to rank and filter molecules from a larger set is to use similarity coefficients. One such way is by using Tanimoto. The experiment below runs similarity tests on a number of molecules using RDKit libraries (http://www.rdkit.org) and Python.
Installing RDKit
To install RDKit on Ubuntu 14.04 desktop OS, do:
sudo apt-get install python-rdkit librdkit1 rdkit-data
If you want to build and install RDKit from source, follow http://www.blopig.com/blog/2013/02/how-to-install-rdkit-on-ubuntu-12-04/.
Similarity
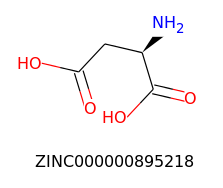
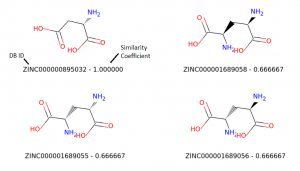
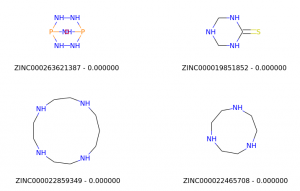
For this experiment, we will use Zinc15 database (http://zinc15.docking.org/) to download a list of molecules in SMILES format, create their respective Morgan fingerprint as a sequence of 1s and 0s, and compare them to a hypothetical molecule to find similar ones using Tanimoto’s similarity.
The Code
The code listing given below is available on GitHub (https://github.com/jod75/chemoinformatics/blob/master/src/SimilarityTest.py).
#!/usr/bin/env python ############################################################################### # Tanimoto Similarity test using RDKit and Zinc15 database # Joseph D'Emanuele # import urllib import os from rdkit import Chem from rdkit.Chem import AllChem from rdkit import DataStructs from rdkit.Chem import Draw ############################################################################### # This runs through the molecules in a SMILES file and returns # a list of Molecules. def process_smiles_file(filename): smiles = {} with open(filename, "r") as infile: infile.readline() # skip header for line in infile: parts = line.split() m = Chem.MolFromSmiles(parts[0]) if m is None: continue smiles[parts[1]] = m return smiles ############################################################################### # similarity test ############################################################################### # create folders if necessary if not os.path.exists("../data"): os.makedirs("../data") if not os.path.exists("../out"): os.makedirs("../out") # download smiles file urllib.urlretrieve("http://files.docking.org/2D/AA/AAAA.smi", "../data/AAAA.smi") # process file and create a list of Molecules molecules = process_smiles_file("../data/AAAA.smi") # use first molecule as query fingerprint fp_query = AllChem.GetMorganFingerprintAsBitVect(molecules[molecules.keys()[0]], 2) # dictionary to keep similarity index similarities = {} # compute Tanimoto similarity for all molecules in our file for moleculeKey in molecules.keys(): fp2 = AllChem.GetMorganFingerprintAsBitVect(molecules[moleculeKey], 2) similarity = DataStructs.FingerprintSimilarity(fp_query, fp2) similarities[moleculeKey] = similarity # get top 20 similar molecules top20 = sorted(similarities, key=similarities.get, reverse=True)[:20] top20.insert(0, molecules.keys()[0]) # this is the query molecule # get bottom 20 similar molecules bottom20 = sorted(similarities, key=similarities.get, reverse=False)[:20] bottom20.insert(0, molecules.keys()[0]) # this is the query molecule # draw top20 similar molecules img = Draw.MolsToGridImage([molecules[x] for x in top20], molsPerRow=2, subImgSize=(400, 400), legends=["%s - %f" % (x, similarities[x]) for x in top20]) img.save("../out/similarities_top20.png") # draw bottom20 similar molecules img = Draw.MolsToGridImage([molecules[x] for x in bottom20], molsPerRow=2, subImgSize=(400, 400), legends=["%s - %f" % (x, similarities[x]) for x in bottom20]) img.save("../out/similarities_bottom20.png")
Sample Output
The following images are sample output from the code above.
Query
Top matches
No match
Thanks for reading this post and happy screening.